FDA 21 CFR Part 11 is the federal regulation that defines how pharmaceutical, biotech, and medical device companies must manage electronic records and electronic signatures. If your maintenance software captures electronic data that the FDA could audit — calibration logs, equipment qualification records, work order histories — your system must meet Part 11 requirements or you risk warning letters, import alerts, and costly consent decrees.
This guide walks you through every requirement in plain language, what a Part 11-compliant CMMS looks like in practice, and how to build a compliance program that survives an FDA inspection.

21 CFR Part 11 was issued by the U.S. Food and Drug Administration in 1997 to set the rules for using electronic records and electronic signatures in place of paper ones. Before Part 11, FDA-regulated companies had to maintain physical paper records signed in ink. The rule opened the door to fully digital operations — but only for companies that could prove their electronic systems are trustworthy, reliable, and equivalent to paper.
The regulation applies to any company that falls under FDA jurisdiction: pharmaceutical manufacturers, biotech firms, medical device makers, food processors under 21 CFR Parts 110 and 117, and compounding pharmacies. If your records support a product that the FDA regulates, Part 11 most likely applies to the systems that create, modify, archive, or transmit those records.
The regulation breaks into two main areas. Subpart B covers electronic records — how you create, store, protect, and retrieve them. Subpart C covers electronic signatures — what makes a digital signature legally binding under FDA rules. Both areas come with specific technical and procedural controls that your systems and people must satisfy.

The FDA's requirements fall into several categories. Each one has direct implications for the software platforms and operational procedures you put in place.
Any system that creates or modifies Part 11 records must generate a computer-generated, time-stamped audit trail. The audit trail must capture who made a change, what the original value was, what it was changed to, and when. Critically, operators cannot modify or delete audit trail entries — only authorized personnel can review them, and only through controlled processes.
In a maintenance context, this means your work order management software must log every status change, every field edit, and every approval action without exception.
Systems must limit access to authorized individuals only. This requires unique user IDs, strong password policies, and role-based permissions that prevent users from seeing or editing records outside their function. Shared logins are a direct Part 11 violation — each person must have their own credentials that are never shared.
Cryotos's user role level access module lets you define exactly who can create, edit, approve, and close records across every department and facility.
An electronic signature under Part 11 must be linked to its associated record in a way that makes it tamper-evident. The regulation requires that signatures include the signer's printed name, the date and time of signing, and the meaning of the signature (such as "reviewed," "approved," or "verified"). Each signing event must require re-authentication — a username and password entry — so that no one can accidentally or intentionally sign a record without deliberate action.
This is where many companies stumble. The FDA requires that any computerized system used to create or maintain Part 11 records be validated — meaning you must formally document that the system does what it is supposed to do, consistently, under all expected conditions. Validation typically involves Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) protocols.
System validation is an ongoing obligation, not a one-time event. Any software update, configuration change, or infrastructure migration that could affect Part 11 records requires re-validation or at minimum a documented impact assessment.
Electronic records must be retained for at least as long as the paper records they replace, and they must be retrievable in human-readable form throughout the retention period. Backing up to a format that requires obsolete software to read is not acceptable — your archive strategy must ensure records stay accessible.
Part 11 is not purely technical. The FDA also requires documented procedures covering how people use regulated systems, how you handle system failures, and how you manage user accounts. Training records showing that users understand Part 11 obligations are routinely requested during inspections.
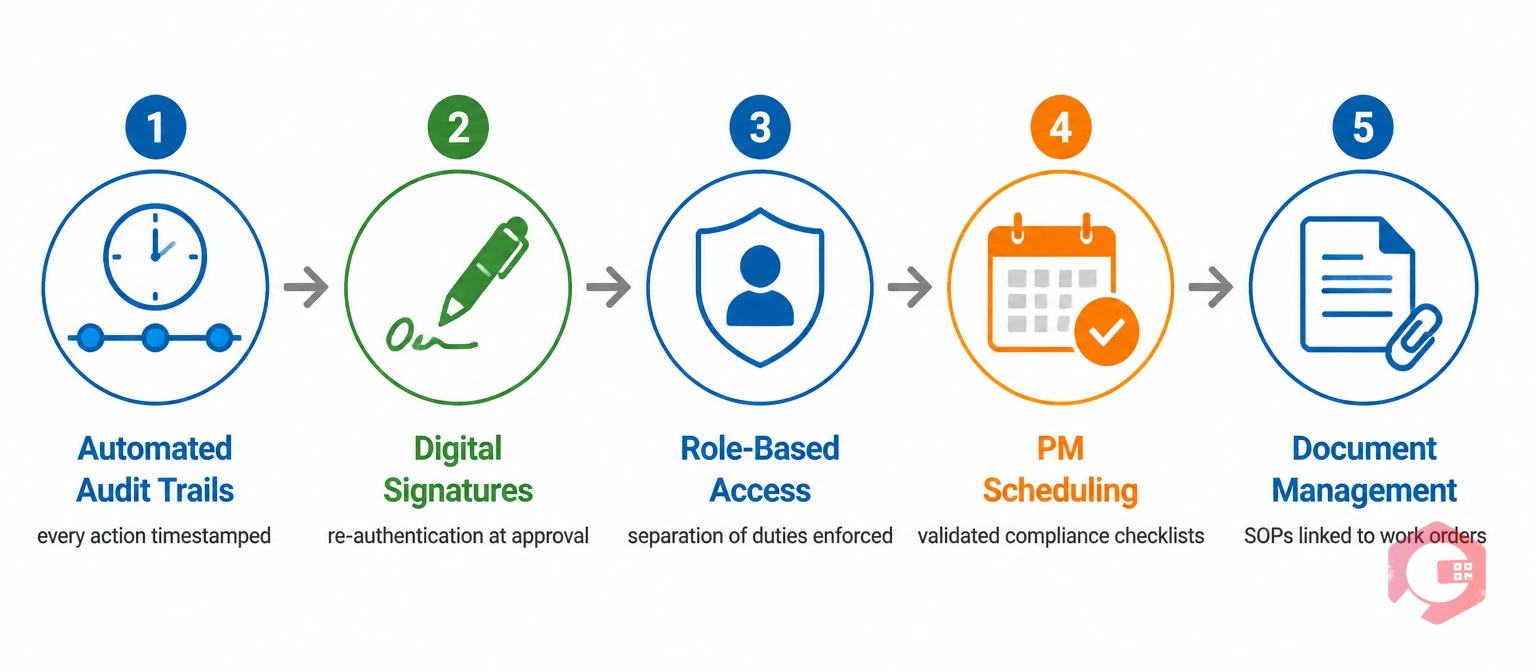
A pharmaceutical manufacturing CMMS sits at the intersection of maintenance operations and regulatory compliance. Calibration records, equipment cleaning logs, preventive maintenance completions, and corrective action records all qualify as Part 11 records when they support a regulated process. Here is how each CMMS capability maps to Part 11 controls.
Every action taken in Cryotos — assigning a technician, closing a work order, updating a checklist item — is timestamped and attributed to the logged-in user automatically. The audit trail is system-generated and cannot be altered by field users. This satisfies the core Part 11 requirement for audit trail integrity without requiring extra manual steps from your team.
Cryotos supports electronic sign-offs on work orders and maintenance checklists that capture the user's identity and timestamp at the point of approval. Each signature event is tied to the user's active session and the specific record being approved, creating the traceability the FDA expects.
You can configure Cryotos so that the technician who performs a task cannot be the same person who approves it. This separation of duties is a common expectation in pharmaceutical GMP environments and aligns directly with Part 11's access control requirements.
Equipment used in GMP manufacturing must be maintained on a documented, verified schedule. Cryotos's preventive maintenance software lets you build validated PM schedules with step-by-step checklists, mandatory fields, and approval workflows that ensure no step is skipped and every completion is recorded with a digital signature.
Part 11 records often need to reference supporting documents — standard operating procedures, calibration certificates, equipment qualification reports. Cryotos's document management feature lets you attach controlled documents directly to assets, work orders, and PM tasks so that inspectors can trace every maintenance action back to its governing procedure.
Use this checklist to assess your current state and build your compliance roadmap. Each item maps to a specific Part 11 requirement
The FDA's Form 483 inspection observations related to electronic records follow predictable patterns. Understanding the most frequent findings helps you close gaps before an inspector does.
This is the single most cited Part 11 finding. Some systems have audit trail capability that is disabled by default or only partially configured. Before relying on any system for Part 11 records, verify that audit trail logging is active for every relevant field — not just select ones.
Manufacturing environments often develop informal practices like shared "line operator" logins because individual account management feels cumbersome. The FDA treats shared accounts as a direct Part 11 violation because they break the traceability requirement. Invest in streamlined onboarding processes so individual accounts are easy to create and maintain.
Many companies perform validation activities but store the documentation loosely. Inspectors will ask to see validation protocols, test scripts, and summary reports for every system in scope. A well-organized validation package that you can retrieve in minutes signals a mature compliance program.
A digital signature that merely captures a typed name without re-authentication does not meet Part 11. Similarly, a signature that does not record the time and the meaning of the signing action is incomplete. Review your current signature workflows against the full definition in 21 CFR 11.50 before your next inspection.
The FDA expects written SOPs for how people use regulated systems, not just technical configurations. If your workflow automation software is configured correctly but you have no SOP describing the process, that gap will appear in your 483 observations.
If your company operates in both the U.S. and European markets, you will also need to satisfy EU GMP Annex 11, the European equivalent of Part 11. The two frameworks are broadly aligned but differ in several important ways.
If you are building a compliance program for both jurisdictions, design to Annex 11 — it is generally the stricter standard, and satisfying it will cover the Part 11 requirements as well.
Compliance is not a project you finish — it is a program you run. The companies that pass FDA inspections consistently are the ones that have built compliance into their day-to-day operations rather than treating it as an audit-prep exercise.
Designate a Part 11 owner for each system in scope. This person is responsible for keeping the validation current, managing the access control list, and reviewing audit trail reports on a defined schedule. Without clear ownership, compliance work falls through the cracks between IT, quality, and operations.
Every software update, infrastructure change, or configuration modification to a Part 11 system should automatically trigger a compliance review. Build this into your change control SOP so it happens by default, not as an afterthought.
Cryotos's report builder lets quality teams schedule automated reports that surface anomalies — unsigned work orders, overdue PMs, unauthorized access attempts — before an inspector finds them. Proactive monitoring is far less disruptive than reactive remediation after a 483 observation.
Part 11 awareness needs regular reinforcement. Annual refresher training, change-specific training when systems are updated, and documentation of every training event will demonstrate to inspectors that your program is active and effective.
Yes, if those maintenance records support a regulated process. Calibration records, equipment qualification logs, cleaning verification records, and preventive maintenance completions for GMP equipment all qualify as Part 11 records when they are created, modified, and stored electronically. The test is whether the record is required by an FDA predicate rule — if it is, and you keep it electronically, Part 11 applies.
Consequences range from Form 483 observations (which require a written response and corrective action plan within 15 business days) to Warning Letters, which are publicly posted and can block product approvals. Serious or repeated violations can lead to consent decrees, import alerts, or facility shutdowns. The financial and reputational costs of non-compliance are far higher than the investment in a proper compliance program.
Cloud-based CMMS software can be fully Part 11 compliant if it provides the required technical controls: system-generated audit trails, role-based access, electronic signature functionality, and a supplier-provided validation package. You should also execute a quality agreement with the vendor that defines each party's responsibilities for maintaining the system in a validated state.
Retention periods are set by the predicate rules, not by Part 11 itself. For example, batch production records under 21 CFR Part 211 must be retained for at least three years after the batch's distribution date or one year after the product's expiration date, whichever is later. Your retention schedule should be built from the specific regulations that govern each record type.
A predicate rule is any existing FDA regulation that requires you to create and maintain a specific record — for example, the GMP regulations in 21 CFR Parts 210 and 211 require batch records, equipment logs, and training records. Part 11 sets the standards for those records when you choose to keep them electronically instead of on paper. Part 11 does not create new record-keeping obligations — it regulates the electronic format of records already required by the predicate rules.
21 CFR Part 11 compliance is achievable without turning your maintenance operations into a paperwork exercise. The key is choosing systems that build the required controls in by default — audit trails that run automatically, access controls that enforce separation of duties, and digital signature workflows that satisfy the FDA's re-authentication requirement without slowing your technicians down.
Cryotos is built for regulated industries. Whether you manage pharmaceutical manufacturing, biotech, or food processing facilities, Cryotos gives your quality and maintenance teams the tools to stay inspection-ready every day. Explore Cryotos and see how a modern CMMS can make Part 11 compliance part of your daily workflow rather than a periodic scramble before an audit.




Cryotos AI predicts failures, automates work orders, and simplifies maintenance—before problems slow you down.












