
21 CFR Part 11 compliance is the FDA regulation that governs how pharmaceutical, biotech, and medical device manufacturers create, sign, and maintain electronic records and electronic signatures — ensuring they carry the same legal weight as paper-based records. Any manufacturer using a computerised system to generate, modify, maintain, archive, retrieve, or transmit records that FDA regulations require must comply with Part 11. Non-compliance can trigger FDA warning letters, consent decrees, and — in the most serious cases — product recalls that cost tens of millions of dollars. According to the FDA's guidance on Part 11 scope and application, the regulation is not about the format of the record — it is about the integrity, traceability, and accessibility of that record.
Key Takeaways

Title 21 of the Code of Federal Regulations, Part 11 — commonly written as 21 CFR Part 11 — was finalised by the FDA in 1997 as the first federal rule to explicitly address electronic records and electronic signatures in regulated industries. The regulation sits under 21 CFR Chapter I, Subchapter A, and applies to any record that FDA regulations require you to keep or submit — batch records, maintenance logs, calibration records, equipment cleaning records, deviation reports, CAPA documentation, and any other quality or operational record that falls under a predicate rule such as cGMP (21 CFR Parts 210/211), QSR (21 CFR Part 820), or GLP (21 CFR Part 58).
Part 11 distinguishes between two types of computerised systems, each carrying different compliance obligations. Most internal CMMS platforms qualify as closed systems, but the distinction matters for how you design your compliance controls.
| Dimension | Closed System | Open System |
|---|---|---|
| Definition | Access is controlled by persons responsible for the content of the records | Access is not controlled by those responsible for the records |
| Common examples | Internal CMMS, validated LIMS, in-house quality system | Email, internet-accessible portals, shared external platforms |
| Required controls | Audit trails, access controls, electronic signatures, system validation | All closed-system controls plus encryption and document authentication |
| Regulatory reference | §11.10 | §11.30 |

The regulation is organised around two major areas: electronic records (Subpart B) and electronic signatures (Subpart C). Each carries specific technical and procedural controls that manufacturers must implement and document.
Understanding the requirements is only the first step. Passing an FDA inspection requires a program built on four interconnected pillars. Missing any one of them creates an exposure that a good audit trail alone cannot fix.
Validation is the foundation. Every computerised system subject to Part 11 must be validated before use in production, and revalidated whenever changes could affect its Part 11-relevant functions. The standard approach follows three sequential protocols:
The ISPE GAMP 5 framework is the most widely recognised guidance for pharmaceutical computer system validation. It categorises systems by complexity and provides risk-based validation requirements for each category.
An audit trail that cannot be retrieved, read, or defended in an inspection is worthless. Effective audit trail management requires more than turning on the feature — it requires a documented policy covering what is captured, how long it is retained, who can access it, and how it is reviewed during normal operations. FDA investigators specifically look for evidence that audit trails are reviewed regularly, not just available on demand. The report builder in Cryotos supports scheduled audit trail review reports, creating a documented evidence trail that routine review is occurring.
Access control failures are the most common Part 11 observation cited in FDA warning letters. The most frequent problems are shared user IDs, inactive accounts that remain open, insufficient role-based permissions, and lack of formal procedures for account creation, modification, and termination. A strong program requires technically enforced role-based access — not just a policy that says users should not share credentials. User role level access controls built into the CMMS prevent unauthorised actions at the system level, regardless of what users might otherwise attempt.
Technical controls alone are not sufficient. Part 11 explicitly requires written policies governing the use of the system, the handling of electronic records, and the management of electronic signatures. Every person who creates, signs, or manages required records must be trained on those procedures — and that training must be documented. According to the FDA's inspection guidance for computerised systems, investigators routinely ask to see training records for system users as part of any Part 11 inspection.
If your pharmaceutical manufacturing CMMS is not yet architected to support all four pillars, now is the time to close those gaps — before your next inspection finds them.
FDA warning letters and 483 observations related to Part 11 cluster around five recurring failures. Knowing them in advance is the most efficient way to close gaps before an inspection finds them.
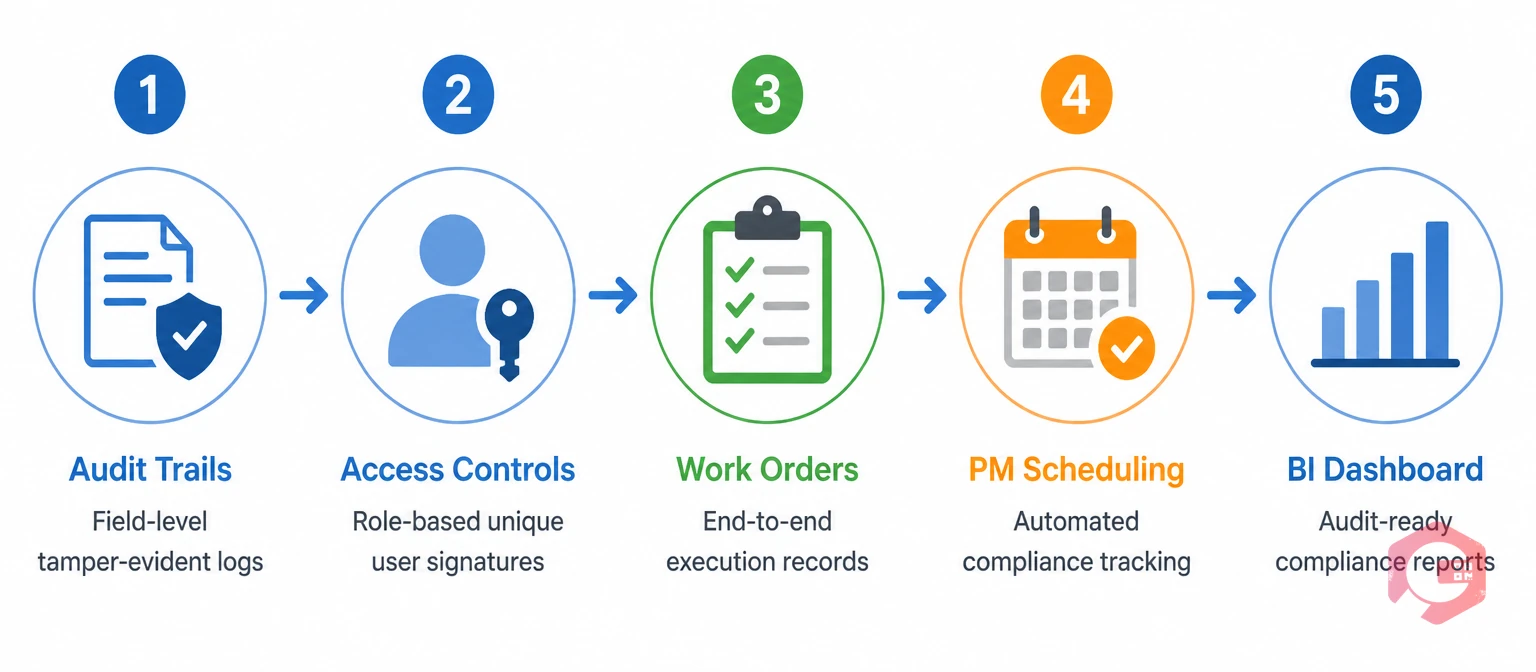
A CMMS is one of the most critical systems for Part 11 compliance in pharmaceutical manufacturing because it generates many of the records that FDA predicate rules require — equipment maintenance logs, calibration records, cleaning records, and PM completion records. A CMMS architected for Part 11 removes much of the manual burden and human error risk from these requirements. Here is how each major requirement maps to CMMS functionality.
Every creation, modification, or deletion of a maintenance record in Cryotos is automatically captured in a tamper-evident, time-stamped audit log. The log records the user who performed the action, the exact field that was changed, the old value, the new value, and the timestamp — at the field level, not just at the record level. Field-level granularity is what Part 11 requires and what FDA investigators look for when reviewing audit trail functionality during inspections.
Cryotos enforces role-based access controls at the system level. User accounts are unique to each individual, credentials cannot be shared, and permissions are configured by role to restrict access to only the functions each user type requires. When a maintenance technician completes a work order or a quality manager approves a deviation, the electronic signature is captured with the user's identity, timestamp, and the meaning of the signature — completion, review, or approval. The maintenance checklists module enforces step-by-step sign-off at the task level, creating a granular execution record.
Maintenance and calibration work orders in Cryotos capture every step of the execution process — task completion, parts used, actual start and end times, technician ID, attachments, and deviations from standard procedure. The work order management module creates an end-to-end record that satisfies both the cGMP record-keeping requirements of 21 CFR Part 211 and the electronic record integrity requirements of Part 11 simultaneously.
The preventive maintenance software in Cryotos schedules equipment PMs based on time intervals, usage meters, or condition triggers. Each scheduled PM generates a work order automatically, and the system records whether each PM was completed on time, late, or missed entirely — creating the PM compliance rate data FDA investigators review to assess whether a manufacturer's maintenance programme is effective.
SOPs, calibration procedures, and equipment manuals attach directly to equipment records and work orders through the document management feature. Technicians access the current approved version of a procedure directly from the work order on their mobile device — eliminating the version control risk that arises when printed SOPs are used on the shop floor.
The BI Dashboard tracks PM compliance rates, overdue work orders, calibration expiry dates, and open deviations in real time. QA and maintenance teams generate audit-ready reports on demand, filtered by date range, asset, department, or user — reducing pre-inspection preparation from days to minutes.
Use this checklist to assess your current compliance status against the regulatory compliance checklist. Each gap you identify represents a potential FDA observation — address them before your next inspection, not during it.
Part 11 applies to any person who, in fulfilment of a requirement in an FDA regulation, uses electronic records in place of paper records or uses electronic signatures. If your facility is subject to any FDA predicate rule — including cGMP under 21 CFR Parts 210 and 211 — and you use any computerised system to maintain the records those rules require, Part 11 applies to those systems. The scope is based on whether you are using an electronic record to fulfil an FDA requirement, not on the size or type of your facility.
Both regulations address computerised systems and electronic records in the pharmaceutical industry, but they originate from different regulatory bodies. 21 CFR Part 11 is a US FDA regulation; EU Annex 11 is a European GMP requirement. The two share significant overlap — particularly around validation, audit trails, access controls, and electronic signatures — but EU Annex 11 has a stronger explicit risk management emphasis and provides more detailed guidance on supplier and service provider management.
Using a cloud-based CMMS does not eliminate your Part 11 obligations — it transfers some technical controls to the vendor but does not transfer compliance responsibility. You remain responsible for ensuring the vendor's system is validated, audit trails function correctly in your configuration, and access controls are implemented appropriately. Your supplier qualification programme must include an assessment of the vendor's quality system, and you should obtain a vendor validation documentation package as part of your compliance evidence.
There is no fixed revalidation interval in Part 11. Revalidation is required when changes are made to the system — software upgrades, configuration changes, or changes to the hardware or operating environment — that could affect Part 11-relevant functions. Best practice is a change control process that identifies which changes require revalidation, defines the scope, and documents the results. Annual routine revalidation reviews are also common even in the absence of specific changes, as documented evidence that the system remains in a validated state.
FDA investigators typically request the system inventory, validation documentation, audit trail samples for specific records, user access lists showing active accounts and role assignments, training records, change control records, and backup and recovery test results. They frequently test audit trail functionality directly by making changes in the system and verifying those changes are captured correctly. They also look for evidence of routine audit trail review — not just the existence of the audit trail functionality.
21 CFR Part 11 compliance is an ongoing operational discipline, not a one-time project. The manufacturers who consistently pass FDA inspections treat Part 11 as part of their normal quality management system — not as a separate exercise revisited only before an audit. Schedule a free demo to see how Cryotos supports a defensible, audit-ready Part 11 programme — from field-level audit trails and role-based access controls to PM compliance reporting and electronic signature capture.




Cryotos AI predicts failures, automates work orders, and simplifies maintenance—before problems slow you down.











