Kaizen in medical equipment manufacturing is a disciplined approach to continuous, incremental improvement applied across production, quality, maintenance, and compliance processes — with every change validated against regulatory requirements before being standardized. In a sector where a single defect can mean a product recall, a failed audit, or direct harm to a patient, Kaizen gives manufacturers a structured way to improve without introducing new risk.
Medical device companies such as Medtronic, Stryker, and GE HealthCare have embedded Kaizen into their manufacturing systems precisely because it aligns with FDA 21 CFR Part 820 and ISO 13485 requirements for continuous improvement within a Quality Management System (QMS). The methodology doesn’t just make processes more efficient — it makes them more auditable, more repeatable, and more defensible under regulatory scrutiny.
Key Takeaways

Kaizen — from the Japanese words “kai” (change) and “zen” (good) — means structured, continuous improvement through small, incremental changes made by the people closest to the work. In general manufacturing, that might mean reorganizing a workbench or trimming two steps from a packaging process. In medical equipment manufacturing, it means doing exactly that — but only after completing a change control assessment, updating the relevant SOPs, and getting sign-off from quality engineering.
That distinction matters. The regulated nature of medical device manufacturing means Kaizen cannot be informal. The FDA’s Quality System Regulation and ISO 13485 both require documented evidence of continuous improvement within the QMS. Kaizen, when implemented correctly, doesn’t just satisfy that requirement — it creates the paper trail that proves it.
At its core, Kaizen in this sector focuses on four principles:

Medical device manufacturers operate under a set of pressures that make continuous improvement both more necessary and more difficult than in other industries. Regulatory requirements from the FDA, ISO, and regional bodies demand documented evidence that quality processes are actively improving — not just stable. At the same time, the cost of getting it wrong is high: defect-related recalls in the medical device sector cost an average of $600 million per incident according to data from the FDA Medical Device Recall Database.
Kaizen addresses the core tension between improving efficiency and maintaining compliance. Because changes are small, controlled, and validated before standardization, Kaizen reduces the risk profile of each individual improvement compared to large-scale process redesigns. The cumulative effect — dozens of validated small improvements per year — produces significant competitive advantage without the regulatory exposure of a major system overhaul.
The five areas where Kaizen produces the highest return in medical manufacturing are consistent across device types, facility sizes, and regulatory frameworks:
Quality is non-negotiable in medical device manufacturing, but that doesn’t mean quality processes can’t be improved. Kaizen teams that focus on reducing the time from defect detection to corrective action closure consistently find significant efficiency gains without compromising rigor.
Common improvements in quality systems include:
A targeted Kaizen improvement in CAPA effectiveness is one of the highest-value activities in regulated manufacturing because it reduces both defect costs and the regulatory burden of managing open corrective actions.
Cleanroom environments are among the most expensive spaces in any manufacturing facility to operate. Labor, environmental control, gowning time, and contamination risk all add cost to every minute of production. Kaizen teams in cleanroom settings focus on reducing non-value-adding activity without introducing contamination risk.
Common improvements in cleanroom environments typically address:
Each of these changes must be validated in the cleanroom context — confirming that the new layout or process doesn’t introduce new contamination pathways before it becomes the standard.
Production equipment in medical device manufacturing isn’t just a productivity asset — it’s a validated system. Equipment downtime doesn’t just slow output; it can trigger re-validation activities, create gaps in batch records, and delay product release. Applying this methodology to maintenance processes reduces both the frequency and the duration of equipment-related disruptions.
Common improvements in equipment management typically include:
Teams using a CMMS to manage equipment maintenance data report up to 30% reductions in unplanned downtime when Kaizen improvements are grounded in actual failure history rather than intuition. The CMMS provides the data foundation — Kaizen provides the methodology for acting on it.
Documentation accuracy is a compliance requirement in medical device manufacturing, not an optional quality measure. FDA 21 CFR Part 820 and ISO 13485 both require complete, accurate records for every production batch, inspection event, and quality decision. Kaizen teams that focus on documentation processes reduce both the error rate and the administrative burden of compliance.
High-impact documentation improvements include:
Every documentation Kaizen improvement has a dual benefit: it reduces the internal cost of compliance administration and strengthens the audit trail that regulators examine during inspections.
Medical device components are often expensive, shelf-life-sensitive, and subject to supplier qualification requirements. Excess inventory ties up capital; insufficient inventory stops production lines and creates supply chain risk. This approach finds the balance through data-driven, incremental adjustment.
Practical improvements in inventory management include:
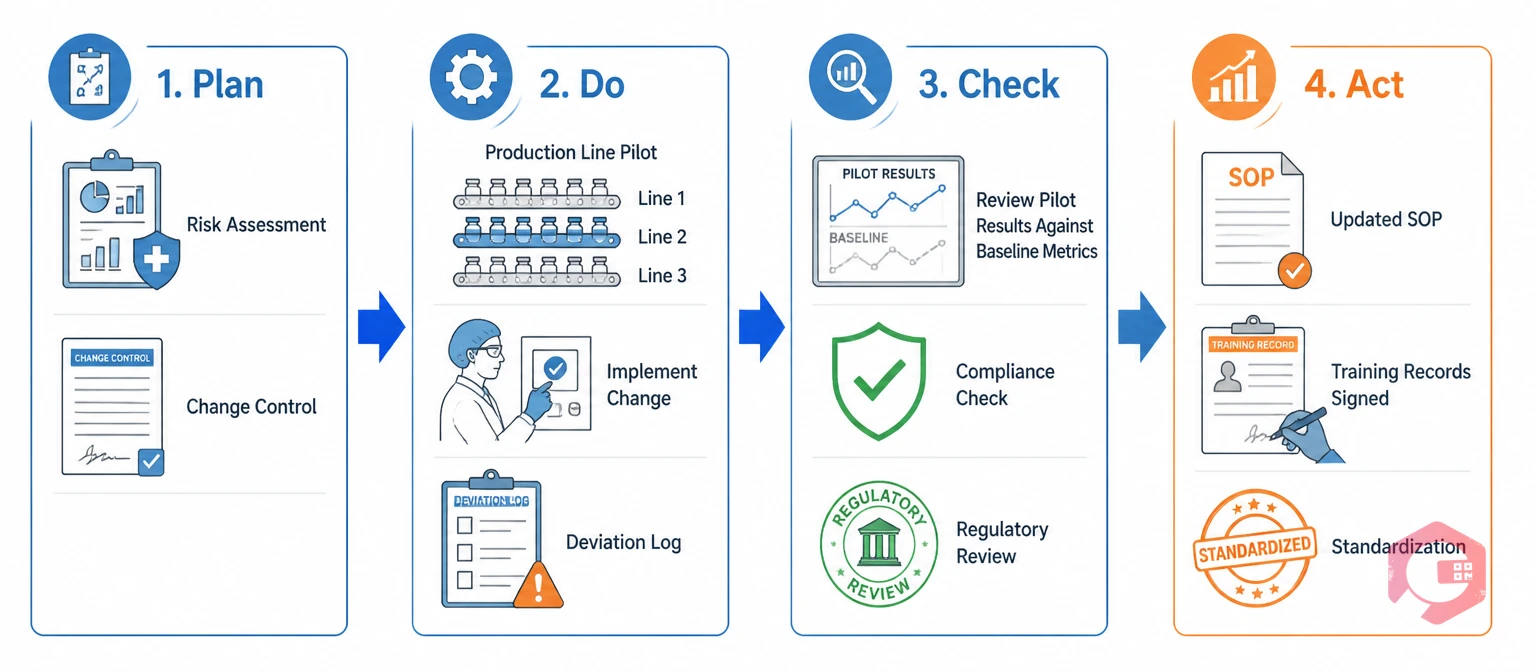
The Plan-Do-Check-Act (PDCA) cycle is the structured framework that ensures every Kaizen improvement in a regulated environment is tested, validated, and standardized before it becomes permanent. Without PDCA, Kaizen in medical manufacturing risks creating undocumented process changes — a direct compliance violation.
Each phase of the cycle carries specific responsibilities in a regulated context:
The regulated PDCA cycle is slower than its general manufacturing equivalent — and intentionally so. The additional validation and documentation steps are what make Kaizen improvements defensible during an FDA inspection or ISO audit. According to the ISO 13485:2016 standard, a medical device QMS must include processes for monitoring, measurement, analysis, and improvement — PDCA-driven Kaizen satisfies all four requirements simultaneously.
Not every improvement opportunity in medical manufacturing calls for the same type of Kaizen activity. Three formats serve different time horizons and problem types:
Kaizen improvements must produce measurable, documented results to satisfy both internal performance goals and regulatory requirements for continuous improvement evidence. The metrics that matter most in medical device manufacturing align directly with quality, efficiency, and compliance outcomes:
| Metric | What It Measures | Why It Matters in Medical Manufacturing |
|---|---|---|
| First Pass Yield (FPY) | Percentage of units completing production without rework | Directly measures quality improvement and reduces rework cost |
| CAPA Cycle Time | Days from defect identification to corrective action closure | Open CAPAs are a primary finding in FDA inspections |
| Equipment Uptime / OEE | Overall Equipment Effectiveness across production assets | Tracks reliability improvements from maintenance Kaizen events |
| Audit Observation Rate | Number of findings per internal or external audit | Declining trend is direct evidence of continuous improvement for regulators |
| Defect Escape Rate | Defects reaching downstream customers or patients | The ultimate quality measure in medical device manufacturing |
| Change Control Cycle Time | Time to complete documentation for process improvements | Faster change control enables faster Kaizen implementation |
Tracking these metrics before and after each Kaizen event creates the documented evidence of improvement that ISO 13485 and FDA QSR require. It also gives Kaizen teams the quantitative feedback loop that drives the next cycle of improvement.

Kaizen is not difficult in concept, but medical manufacturing creates specific implementation challenges that general Kaizen literature doesn’t fully address. The most common failure modes are:

Individual Kaizen events produce measurable gains; a Kaizen culture produces compounding gains that become a sustainable competitive advantage. Building that culture in a regulated environment requires deliberate investment in four areas:
Kaizen in medical manufacturing depends on reliable data — failure frequencies, equipment uptime, maintenance cycle times, inventory consumption rates. Cryotos healthcare CMMS gives maintenance and quality teams the real-time visibility they need to identify improvement opportunities, measure the impact of changes, and demonstrate continuous improvement to regulators.
Specifically, Cryotos supports the Kaizen cycle through:
Kaizen focuses on frequent, small, incremental improvements driven by the people doing the work. Six Sigma is a structured statistical methodology for reducing process variation and defects, typically applied to larger, more complex problems. In medical manufacturing, the two are often used together: Kaizen for ongoing daily and weekly improvements, Six Sigma for tackling high-impact quality problems that require deep statistical analysis. Both require change control documentation in a regulated environment.
It depends on the nature of the change and how the process is validated. Changes to validated processes that affect product quality, safety, or efficacy typically require a completed change control record and, depending on the change type, may require a new validation study. Minor changes to non-product-contact processes or administrative procedures typically do not require FDA notification. Your QMS change control procedure governs which changes require which level of review — and all Kaizen improvements should flow through that procedure without exception.
There is no single right number — it depends on team size, manufacturing complexity, and improvement maturity. Early-stage Kaizen programs typically run 2–4 formal Kaizen Blitz events per year per production area, alongside daily Kaizen activities embedded in standard operations. Mature programs may run higher volumes of smaller, faster improvement cycles. The quality of implementation and the discipline of standardization matter more than event frequency.
FDA auditors examining a medical device QMS under 21 CFR Part 820 look for documented evidence that the quality system is producing measurable improvement over time. The strongest evidence includes trending defect rate data showing a declining pattern, CAPA cycle time data showing faster resolution, First Pass Yield improvements tied to specific corrective actions, and audit observation trend data showing fewer findings per audit cycle. Kaizen event records — including baseline data, change descriptions, pilot results, and standardization documentation — directly support all four of these evidence types.
If your team is building a Kaizen program that needs the data infrastructure to measure, track, and demonstrate continuous improvement, schedule a free demo to see how Cryotos CMMS supports medical equipment manufacturers with real-time equipment data, maintenance tracking, and audit-ready records.




Cryotos AI predicts failures, automates work orders, and simplifies maintenance—before problems slow you down.












